Новый вариант SARS-CoV-2, выявляемый в Великобритании

Оригинал: cogconsortium.uk, gov.uk, virological.org
Авторы: 1-я статья — The COVID-19 Genomics UK (COG-UK) consortium, 2-я статья — gov.uk, 3-я статья — Andrew Rambaut et al.
Опубликовано: cogconsortium.uk 14.12.2020 г., gov.uk 14.12.2020 г., virological.org 19.12.2020 г.
Переводчик-Модератор: Анна Селюта, Фонд медицинских решений «Не напрасно!»
Переводчики: Анна Селюта, Фонд медицинских решений «Не напрасно!», Ирина Лагерь, Фонд медицинских решений «Не напрасно!», Снежанна Генинг, Фонд медицинских решений «Не напрасно!»
Редакторы: Наталья Рожкова, Дарья Цыба, Саша Васильева, Фонд медицинских решений «Не напрасно!»
1. Что такое «новый вариант» SARS-CoV-2, выявляемый в Великобритании?
Вариант, описанный сегодня в Палате общин Великобритании, содержит новый набор мутаций, связанных с вариантом, быстро распространяющимся на юго-востоке Англии (и в более широком смысле), которая является предметом текущих исследований Агентства общественного здравоохранения Великобритании, координируемых Общественным здравоохранением Англии при поддержке COG-UK. Этот вариант несет набор мутаций, включая мутацию N501Y в мотиве связывания рецептора спайк-белка, который вирус использует для связывания с человеческим рецептором ACE2 (АПФ2, ангиотензинпревращающего фермента 2 — прим.перев.).
Этот вариант был выявлен благодаря активному и усиленному мониторингу общественного здравоохранения Англии (Public Health England’s — PHE) после увеличения числа случаев заболевания в Кенте и Лондоне. Вариант получил название «VUI - 202012/01» (первый вариант находится на рассмотрении в декабре 2020 года).
По состоянию на 13 декабря было выявлено 1108 случаев этого варианта, преимущественно на юге и востоке Англии. PHE работает с партнерами над расследованием и планирует поделиться своими выводами в течение следующих 2-х недель. Большое количество случаев этого варианта вируса наблюдалось в некоторых регионах, где также высока заболеваемость COVID-19. Пока неизвестно, является ли этот вариант причиной такого увеличения числа случаев заражений COVID-19. PHE будет отслеживать последствия этого в ближайшие дни и недели.
Вирусы нередко мутируют; сезонный грипп мутирует каждый год. Варианты SARS-CoV-2 наблюдались в других странах, например, в Испании.
Этот вариант включает мутацию в спайк-белке. Изменения в этой части спайк-белка могут привести к тому, что вирус станет более заразным и будет легче распространяться между людьми.
2. Способствует ли выявленная мутация увеличению передачи вируса, более тяжелому протеканию заболевания или неэффективности вакцины?
В настоящее время предпринимаются усилия, чтобы подтвердить, способствуют ли какие-либо из этих мутаций увеличению передачи вируса среди населения. В настоящее время нет доказательств того, что этот вариант вируса (или любой другой, изученный на сегодняшний день) имеет какое-либо влияние на тяжесть заболевания или вакцины против SARS-CoV-2 будут менее эффективными, хотя оба вопроса требуют дальнейших исследований, проводимых в темпе. Мы будем предоставлять дальнейшие обновления по мере продолжения расследования.
В настоящее время нет доказательств, позволяющих предположить, что этот вариант каким-либо образом влияет на тяжесть заболевания, выработку антител или эффективность вакцины.
3. Что известно о мутациях SARS-CoV-2?
Мутации возникают естественным образом в геноме SARS-CoV-2 по мере того, как вирус реплицируется и циркулирует среди людей. Они накапливаются со скоростью от 1 до 2 мутаций в месяц в глобальной филогении (филогенез — развитие биологического вида во времени. прим. перев.). В результате этого продолжающегося процесса с момента появления вируса в 2019 году в геноме SARS-CoV-2 уже возникли много тысячи мутаций. По мере того как мутации продолжают возникать, все чаще наблюдаются новые комбинации.
Подавляющее большинство мутаций, наблюдаемых в SARS-CoV-2, не оказывают видимого воздействия на вирус, и лишь очень малое количество из них, вероятно, будут важны и изменят вирус каким-либо заметным образом (например, изменение способности заражать людей; вызывать заболевание различной степени тяжести; или становиться нечувствительными к воздействию иммунного ответа человека, в том числе ответу, вызываемому вакциной).
Трудно предсказать, насколько важна та или иная мутация, когда она впервые возникает, на фоне непрерывного появления новых мутаций. Понимание их значения может быть возможным на основе экспериментальной работы, которая показывает связь между мутацией и незначительными изменениями в биологии вируса. Однако для проверки эффекта многих тысяч комбинаций мутаций потребовалось бы много времени и усилий. Наибольшую озабоченность вызывают любые изменения, которые приводят к увеличению числа повторных инфекций или неудачной вакцинации (сигнализируя о том, что вирус может уклоняться от иммунной защиты, вызванной предыдущей инфекцией или вакцинацией).
Наибольшее внимание уделяется мутациям в гене, кодирующем спайк-белок, который связан с проникновением вируса в клетку. В настоящее время существует около 4000 мутаций в гене спайк-белка (обратите внимание, что в одной и той же точке генома может произойти другая мутация, поэтому количество мутаций больше, чем фактическое количество оснований в гене спайк-белка). Небольшое количество мутаций происходит в области, называемой рецептор-связывающим мотивом (receptor binding motif — RBM) спайк-белка, который отвечает за проникновение вируса через его взаимодействие с человеческим рецептором (hACE2) в клетках человека.
4. Каким образом ученые отслеживают мутации в геноме SARS-CoV-2?
COG-UK (в число партнеров которой входят четыре агентства общественного здравоохранения) проводит секвенирование случайно выбранных положительных образцов по всей Великобритании и публикует отчет об охвате секвенированием, который направляется в агентства здравоохранения каждую неделю. Краткая версия находится в открытом доступе cogconsortium.uk. Случайность выборки важна для охвата региональных особенностей (секвенирование эпидочагов также важно, но его результаты будут демонстрировать геномы одного и того же варианта вируса, что не способствует идентификации мутировавших вирусов). Общий охват секвенированием в Великобритании составляет около 10% (доля секвенированных образцов от числа всех положительных).
Также COG-UK разработала программное обеспечение для автоматизированного анализа мутаций, например: cov-glue.cvr.gla.ac.uk.
COG-UK отслеживает мутации во времени и пространстве, используя два подхода:
- Статистический подход подразумевает автоматизированный статистический анализ, филогенетические исследования и ежедневное сопоставление результатов работы для создания отчета о типах и частотах мутаций в секвенированной популяции.
- Таргетный подход заключается в поиске и отслеживании мутаций, которые уже были охарактеризованы в лабораторных экспериментах как потенциально важные (например, потому что они могут повлиять на взаимодействие с иммунной системой человека).
COG-UK разработал сводный отчет о мутациях, который будет еженедельно публиковаться в Интернете в качестве дополнения к отчету об охвате секвенированием. Первый прототип выпущен 18 декабря, со временем в него будут вноситься улучшения. Он будет сосредоточен на часто встречающихся и/или важных мутациях. Такие отчеты — предварительные данные, поскольку они будут включать лишь часть от истинного числа случаев.
5. Какие научные данные о новом штамме SARS-CoV-2, выявленном в Великобритании, существуют на сегодняшний день?
Сообщается о быстро распространяющемся штамме вируса в Великобритании, несущем в своём геноме неожиданно большое количество генетических изменений, затрагивающих помимо прочего рецептор-связывающий домен и сайт расщепления фурина. Учитывая (i) предсказанные экспериментально и логичные фенотипические последствия некоторых из этих мутаций, (ii) неизученные эффекты данных мутаций в комбинации, и (iii) высокую скорость распространения штамма B.1.1.7 в Великобритании, этот вариант требует срочного лабораторного исследования и активного генетического тестирования во всем мире.
6. Какова предварительная геномная характеристика нового штамма SARS-CoV-2, выявленного в Великобритании, несущего новый набор мутаций в гене S-белка (спайк-белка)?
Недавно в базе данных COG-UK был обнаружен отдельный филогенетический кластер (названный B.1.1.7). Этот кластер быстро рос в течение последних 4 недель и начал появляться в других районах Великобритании. Некоторые характеристики кластера заслуживают внимания по эпидемиологическим и биологическим причинам.
Линия B.1.1.7 все чаще встречается в регионах Великобритании. Растет как число случаев инфекции, так и число вовлеченных регионов. B.1.1.7 имеет необычайно высокое число генетических мутаций, в частности, в гене спайк-белка.
Три из этих мутаций имеют потенциальные биологические эффекты, которые были описаны ранее в экспериментах:
- Мутация N501Y в одном из шести ключевых контактных аминокислотных остатков рецептор-связывающего домена (RBD) повышает аффинность к человеческому и мышиному АПФ2.
- Делеция гена спайк-белка 69-70del описана в контексте ускользания вируса от иммунного ответа у человека, а также в связи с другими изменениями RBD.
- Мутация P681H находится в непосредственной близости к сайту расщепления фурина (фурин — фермент клеток человека, активность которого необходима для размножения вируса и инфицирования новых клеток — прим.ред.).
Быстрый рост числа случаев указывает на необходимость более активного генетического тестирования и эпидемиологического наблюдения во всем мире и проведения лабораторных исследований антигенности и инфекционности данного штамма.
7. Где именно сейчас обнаруживают новый штамм SARS-CoV-2?
Два самых ранних отобранных генома, принадлежащих к линии B.1.1.7, были собраны 20 сентября 2020 года в Кенте, а другой — 21 сентября 2020 года в Большом Лондоне (округ). Штаммы B.1.1.7 продолжали обнаруживать в Великобритании до начала декабря 2020 года.
Геномы, принадлежащие к линии B.1.1.7, образуют монофилетическую кладу (ветвь потомков), которая хорошо поддерживается большим количеством определяющих линию мутаций. (Рис. 1) На 15 декабря в линии B.1.1.7 насчитывается 1623 генома. Из них 519 были собраны в Большом Лондоне, 555 — в Кенте, 545 — в других регионах Великобритании, включая Шотландию и Уэльс, и 4 — в других странах.
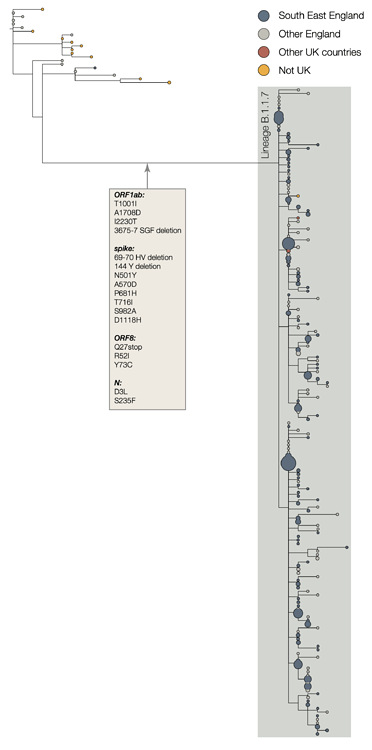
Рис. 1 — Клада B.1.1.7
8. Каковы происхождение мутации и скорость эволюции?
Линия B.1.1.7 несет большее, чем обычно, количество вирусных генетических изменений. Накопление 14 замен аминокислот, специфичных для линии, наблюдается впервые за время пандемии COVID-19. Большинство ветвей глобального филогенетического дерева SARS-CoV-2 показывают не более нескольких мутаций. Они накапливаются с относительно постоянной скоростью. По оценкам, циркулирующие клоны SARS-CoV-2 накапливают примерно по 1–2 нуклеотидные мутации в месяц (Duchene et al. 2020).
Предварительный анализ этих наблюдений показывает регресс генетических расстояний от корня до кончика в зависимости от даты отбора проб генома для линии B.1.1.7 и для выбора родственных геномов внешней группы. Скорость молекулярной эволюции внутри линии B.1.1.7 аналогична скорости эволюции других родственных линий. Однако B.1.1.7 сильнее отклоняется от филогенетического корня пандемии, что указывает на более высокую скорость молекулярной эволюции филогенетической ветви, непосредственно предшествующей B.1.1.7. Кроме того, предполагаемые нуклеотидные изменения в этой ветви преимущественно изменяют аминокислоты (14 несинонимичных мутаций и 3 делеции) на ветке 6 синонимичных мутаций. Это наводит на мысль о процессе, включающем адаптивную молекулярную эволюцию, хотя пока нельзя исключать роль увеличения скорости фиксации за счет ослабления селективных ограничений.
9. Какие эволюционные процессы или селекционный отбор могли привести к появлению B.1.1.7?
О высоких темпах накопления мутаций в течение коротких периодов времени ранее сообщали результаты исследований пациентов с иммунодефицитом или иммуносупрессией, хронически инфицированных SARS-CoV-2 (Choi et al.2020; Avanzato et al.2020; Kemp et al.2020). При этих хронических вариантах детектируемая РНК SARS-CoV-2 обнаруживается в течение 2–4 месяцев или дольше (хотя есть также сообщения о более длительных инфекциях у некоторых иммунокомпетентных людей). Пациентов лечат плазмой выздоравливающих и ремдесивиром. Секвенирование вирусного генома этих инфекций выявляет необычно большое количество нуклеотидных изменений, делеционных мутаций и часто высокое соотношение несинонимичных и синонимичных изменений. Плазма выздоравливающих часто назначается при высокой вирусной нагрузке пациента, и Kemp et al. (2020) сообщают, что генетическое разнообразие вирусов внутри пациента увеличивалось после лечения плазмой.
При таких обстоятельствах ожидается, что эволюционная динамика и давление отбора на популяцию вируса внутри пациента будут сильно отличаться от таковых при типичной инфекции. Во-первых, отбор естественных иммунных ответов у пациентов с иммунодефицитом/подавленным иммунитетом будет слабым или отсутствовать. Во-вторых, отбор, возникающий в результате терапии антителами, может быть сильным из-за высоких концентраций антител. В-третьих, если терапия антителами проводилась после многих недель хронической инфекции, популяция вируса, персистирующего внутри пациента, будет необычно большой и генетически разнообразной. В таких условиях терапия антителами создает условия для фиксации множественных генетических изменений.
Эти соображения приводят нас к гипотезе о том, что необычная генетическая дивергенция линии B.1.1.7 по крайней мере частично могла быть результатом эволюции вируса в хронически инфицированном человеке. Такие инфекции редки, и дальнейшая передача, предположительно, случается еще реже. Но их нельзя исключить, учитывая большое количество новых инфекций.
Хотя мы предполагаем, что хроническая инфекция сыграла роль в происхождении B.1.1.7, мы еще не можем сделать вывод о точной природе этого события.
10. Каково возможное биологическое значение мутаций?
В таблице 1 представлены подробности несинонимичных мутаций и делеций, специфичных для клонов B.1.1.7. Отметим, что многие из них происходят в «шип»-белке вируса. К ним относятся положение «шипа» 501, один из ключевых контактных остатков в рецепторсвязывающем домене (RBD). Экспериментальные данные предполагают, что мутация N501Y может увеличивать сродство к рецептору ACE2 (Starr et al.2020) и P681H — одному из 4 остатков, составляющих вставку, которая создает сайт расщепления фурином между S1 и S2 в «шипе». Сайт расщепления фурином S1/S2 SARS-CoV-2 не обнаружен в близкородственных коронавирусах. Эксперименты на животных моделях показали, что он облегчает проникновение в респираторные эпителиальные клетки и передачу инфекции (Hoffmann, Kleine-Weber и Pöhlmann 2020; Peacock et al. 2020; Чжу и др. 2020). N501Y был связан с повышенной контагиозностью и вирулентностью на модели мышей (Gu et al. 2020). И N501Y, и P681H ранее наблюдались независимо, но, насколько нам известно, возможна их комбинация.
Таблица 1 — Несинонимичные мутации и делеции, предположительно происходящие в ветви, ведущей к линии B.1.1.7.
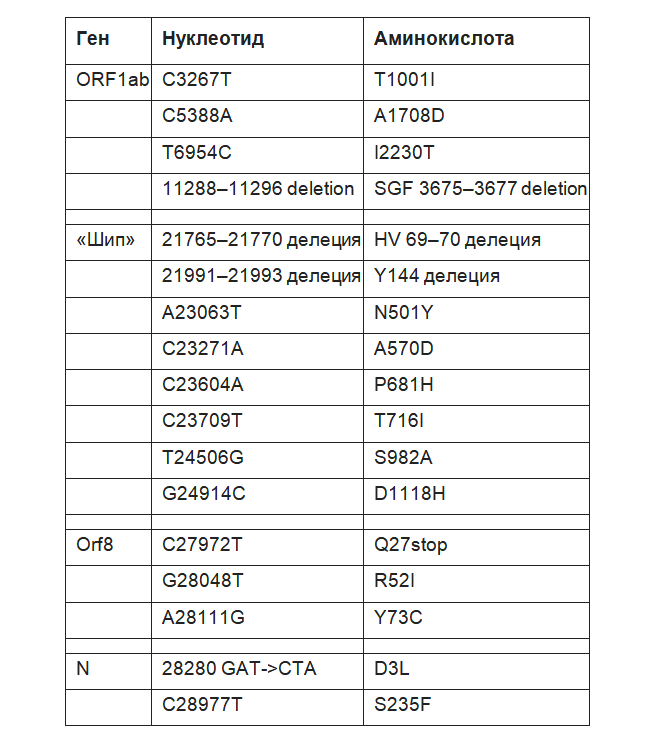
Также присутствует делеция двух аминокислот в сайтах 69–70 в «шипе». Эта мутация — одна из ряда повторяющихся делеций, наблюдаемых в N-концевом домене «шип»-белка (McCarthy et al.2020; Kemp et al.2020) — была замечена в нескольких клонах, связанных с несколькими мутациями RBD. Например, она возникла во время вспышки среди норок в Дании на фоне мутации Y453F RBD, а у людей — в связи с мутацией N439K RBD, что объясняет ее относительно высокую частоту в данных глобального генома (~ 3000 последовательностей).
Вне «шип»-белка мутация ORF8 Q27stop усекает белок ORF8 или делает его неактивным и, таким образом, позволяет накапливаться дальнейшим нижестоящим мутациям. В начале пандемии во всем мире выделили несколько изолятов вируса с делециями, ведущими к потере экспрессии ORF8, включая большой кластер в Сингапуре с делецией. Она приводила как к усеченной экспрессии Orf7b, так и к устранению экспрессии ORF8. Сингапурский штамм с делецией 382nt был связан с более легкой клинической инфекцией и меньшим количеством постинфекционных воспалений, однако этот кластер вымер в конце марта после того, как Сингапур успешно ввел меры контроля (Young et al.2020). Последующая работа показала, что делеция ORF8 оказывает лишь очень умеренное влияние на репликацию вируса в клетках верхних дыхательных путей человека по сравнению с вирусами без делеции. Это приводит к небольшому отставанию репликации по сравнению с вирусами с делецией (Gamage et al.2020). Поскольку ORF8 обычно состоит из 121 аминокислоты, вероятно, стоп-кодон в положении 27, наблюдаемый в линии B.1.1.7, приводит к потере функции.
Наконец, есть 6 синонимичных мутаций с 5 в ORF1ab (C913T, C5986T, C14676T, C15279T, C16176T) и одна в гене M (T26801C).
Литература:
- Avanzato, Victoria A., M. Jeremiah Matson, Stephanie N. Seifert, Rhys Pryce, Brandi N. Williamson, Sarah L. Anzick, Kent Barbian, et al. 2020. “Case Study: Prolonged Infectious SARS-CoV-2 Shedding from an Asymptomatic Immunocompromised Individual with Cancer.” Cell, November. doi.org
- Choi, Bina, Manish C. Choudhary, James Regan, Jeffrey A. Sparks, Robert F. Padera, Xueting Qiu, Isaac H. Solomon, et al. 2020. “Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host.” The New England Journal of Medicine 383 (23): 2291–93. doi.org
- Duchene, Sebastian, Leo Featherstone, Melina Haritopoulou-Sinanidou, Andrew Rambaut, Philippe Lemey, and Guy Baele. 2020. “Temporal Signal and the Phylodynamic Threshold of SARS-CoV-2.” Virus Evolution 6 (2): veaa061. doi.org
- Young, Barnaby E. et al. 2020. “Effects of a Major Deletion in the SARS-CoV-2 Genome on the Severity of Infection and the Inflammatory Response: An Observational Cohort Study.” 2020. The Lancet 396 (10251): 603–11. doi.org
- Gamage, Akshamal M., Kai Sen Tan, Wharton O. Y. Chan, Jing Liu, Chee Wah Tan, Yew Kwang Ong, Mark Thong, et al. 2020. “Infection of Human Nasal Epithelial Cells with SARS-CoV-2 and a 382-Nt Deletion Isolate Lacking ORF8 Reveals Similar Viral Kinetics and Host Transcriptional Profiles.” PLoS Pathogens 16 (12): e1009130. doi.org
- Gu, Hongjing, Qi Chen, Guan Yang, Lei He, Hang Fan, Yong-Qiang Deng, Yanxiao Wang, et al. 2020. “Adaptation of SARS-CoV-2 in BALB/c Mice for Testing Vaccine Efficacy.” Science 369 (6511): 1603–7. doi.org
- Hoffmann, Markus, Hannah Kleine-Weber, and Stefan Pöhlmann. 2020. “A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells.” Molecular Cell 78 (4): 779–84.e5. doi.org
- Kemp, S. A., D. A. Collier, R. Datir, S. Gayed, A. Jahun, M. Hosmillo, Iatm Ferreira, et al. 2020. “Neutralising Antibodies Drive Spike Mediated SARS-CoV-2 Evasion.” Infectious Diseases (except HIV/AIDS). medRxiv. doi.org
- McCarthy, Kevin R., Linda J. Rennick, Sham Nambulli, Lindsey R. Robinson-McCarthy, William G. Bain, Ghady Haidar, and W. Paul Duprex. 2020. “Natural Deletions in the SARS-CoV-2 Spike Glycoprotein Drive Antibody Escape.” Microbiology. bioRxiv. doi.org
- Peacock, Thomas P., Daniel H. Goldhill, Jie Zhou, Laury Baillon, Rebecca Frise, Olivia C. Swann, Ruthiran Kugathasan, et al. 2020. “The Furin Cleavage Site of SARS-CoV-2 Spike Protein Is a Key Determinant for Transmission due to Enhanced Replication in Airway Cells.” Cold Spring Harbor Laboratory. doi.org
- Starr, Tyler N., Allison J. Greaney, Sarah K. Hilton, Daniel Ellis, Katharine H. D. Crawford, Adam S. Dingens, Mary Jane Navarro, et al. 2020. “Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding.” Cell 182 (5): 1295–1310.e20. doi.org
- Zhu, Yunkai, Fei Feng, Gaowei Hu, Yuyan Wang, Yin Yu, Yuanfei Zhu, Wei Xu, et al. 2020. “The S1/S2 Boundary of SARS-CoV-2 Spike Protein Modulates Cell Entry Pathways and Transmission.” Cold Spring Harbor Laboratory. doi.org