Безопасность и эффективность вакцины против COVID-19 на базе векторов rAd26 и rAd5 применяемой в гетерологичном режиме прайм-буст: промежуточный анализ рандомизированного контролируемого клинического испытания III фазы в России

Оригинал: The Lancet
Автор: Logunov et al.
Опубликовано: 02 февраля 2021, The Lancet
Перевод: Полина Дроздова, Фонд медицинских решений «Не напрасно»
Редакция: Наталья Рожкова, Фонд медицинских решений «Не напрасно»
Комментарий: Оксана Станевич, Фонд медицинских решений «Не напрасно», врач-инфекционист клиники ПСПбГМУ им. И.П. Павлова
Комментарий специалиста
Оксана Владимировна Станевич, врач-инфекционист и научный сотрудник ПСПбГМУ им. И.П. Павлова, эксперт "Просто спросить" и "Просто спросить о COVID-19" Фонда "Не напрасно".
В опубликованной статье необходимо принять во внимание несколько важных деталей.
Во-первых, статья является публикацией промежуточного анализа данных III фазы клинического испытания вакцины, что означает вероятное искажение полученных результатов к моменту завершения исследования (апрель-май 2021 года). При этом могут измениться значения эффективности, появиться больше сообщений о нежелательных явлениях, и, самое главное, сообщения о реинфекции после вакцинации (эффективность вакцины в течение времени).
Во-вторых, исследователи не учитывали среди исходов бессимптомные формы заболевания COVID-19, что не дает возможности оценить эффективность вакцины в предотвращении бессимптомных форм инфекции. Это означает необходимость сохранения таких мер предосторожности, как социальное дистанцирование и ношение масок, несмотря на наличие прививки.
В-третьих, чтобы понимать, что такое эффективность вакцины на уровне 91,6%, и иметь возможность сравнивать ее с эффективностями других вакцин, необходимо пересчитать их в единое значение. Таковым может служить отношение рисков, а именно риск заболеть в группе привитых по сравнению с группой плацебо. Так, Спутник V снижает риск заболеть коронавирусной инфекцией в 11,7 раз в группе привитых по сравнению с группой плацебо, Pfizer / BioNTech (Comirnaty) - в 20 раз, Moderna - в 16,7 раз, AstraZeneca/Oxford LD+SD (2х-фазный режим введения, в первый раз - низкая доза, во второй - стандартная доза вакцины) - в 9,8 раз.
Еще важным остается вопрос о том, работает ли вакцина в качестве “экстренной” помощи, когда прививка случилась в инкубационный период COVID-19. Учитывая короткие сроки инкубационного периода этой инфекции (14 дней и меньше), а также довольно долгую выработку антител после введения вакцины (14-21 день), вероятность получить антитела к моменту разгара симптомов - довольно малая. По этой причине рассматривать ее как “экстренную” помощь не следует. Однако, несмотря на это, учитывая высокий риск развития тяжелого и жизнеугрожающего состояния, старайтесь попасть на вакцинацию даже после предполагаемого контакта, особенно если вы относитесь к группе риска.
Не забывайте: в условиях низкой доступности вакцины приоритетными группами для вакцинации будут являться пожилые люди, ослабленные больные, больные с онкологическим диагнозом, а также те, кто с такими людьми проживает или часто контактирует. В первую очередь наша цель защитить эту категорию лиц. Если вам не удается попасть на прививку, мониторьте пункты проведения клинических испытаний - всегда можно стать участником III фазы испытания тех или иных форм и режимов введения разрабатываемых вакцин. Но, если вам доступна вакцинация здесь и сейчас - всегда пользуйтесь этим шансом: опыт показывает, что никогда нельзя предположить, как будет протекать SARS-CoV-2 инфекция даже у молодых людей.
Аннотация
Гетерологичная вакцина Гам-КОВИД-Вак (Спутник V), основанная на рекомбинантном аденовирусе (rAd), показала хороший профиль безопасности и индуцировала сильный гуморальный и клеточный иммунный ответ у участников I-II фаз клинических испытаний. В этой публикации мы представляем предварительные результаты об эффективности и безопасности Гам-КОВИД-Вак на основе промежуточного анализа результатов клинических испытаний III фазы.
Методы
Мы провели рандомизированное двойное слепое плацебо-контролируемое испытание III фазы в 25 больницах и поликлиниках в г. Москве. В испытание включали пациентов старше 18 лет с отрицательными результатами ПЦР-анализа на SARS-CoV-2 и анализов на антитела IgG и IgM, не перенесших инфекционные заболевания в течение 14 дней, предшествующих включению, и не получавших никакие другие вакцины в течение 30 дней, предшествовавших включению. Участники были случайным образом распределены в группы вакцины и плацебо в соотношение 3:1 со стратификацией по возрастной группе.
Исследователи, участники и все сотрудники, принимавшие участие в проведении исследования, не располагали информацией о разделении по группам. Вакцину вводили внутримышечно в количестве 0,5 мл на дозу в режиме прайм-буст (с 21-дневным интервалом между первой дозой (rAd26) и второй дозой (rAd5); оба вектора содержат ген полноразмерного S-гликопротеина SARS-CoV-2). Основным исходом считали долю участников с подтвержденным ПЦР COVID-19 по прошествии не менее чем 21 дня после получения первой дозы. При проведении всех анализов данные участников, в отношении которых был нарушен протокол, исключались. Основной исход рассчитывали у участников, которые получили две дозы вакцины или плацебо; частоту серьезных нежелательных явлений оценивали у всех участников, которые получили хотя бы одну дозу к моменту закрытия базы данных; частоту редких нежелательных явлений оценивали у всех участников, которые получили две дозы и для которых все доступные данные были проверены на момент закрытия базы данных. Клиническое исследование зарегистрировано в системе ClinicalTrials.gov (NCT04530396).
Результаты
В промежутке с 7 сентября по 24 ноября 2020 года 21 977 взрослых участников были случайным образом распределены в группу вакцины (n=16 501) или плацебо (n=5476). 19 866 участников получили две дозы вакцины или плацебо и было включены в анализ основного исхода. Начиная с 21 дня после первой дозы вакцины (день получения второй дозы вакцины) у 16 (0,1%) из 14 964 участников в группе вакцины и 62 (1,3%) из 4902 участников в группе плацебо был подтверждён COVID-19; эффективность вакцины составила 91,6% (95% ДИ 85,6–95,2). Большинство отмеченных нежелательных явлений относились к первой степени (7485 [94,0%] из 7966 событий). 45 (0,3%) из 16 427 участников в группе вакцины и 23 (0,4%) из 5435 участников в группе плацебо столкнулись с серьезными нежелательными явлениями; ни один случай не следует рассматривать как связанный с вакцинацией, что было подтверждено независимой наблюдательной комиссией. В течение исследования были зафиксированы 4 смерти (3 [<0,1%] из 16 427 в группе вакцины и 1 [<0,1%] из 5435 участников в группе плацебо), ни одну из которых не следует считать связанной с вакцинацией.
Интерпретация
Данный промежуточный анализ III фазы клинического испытания Гам-КОВИД-Вак показал 91,6% эффективность против COVID-19 и хорошую переносимость в большой когорте.
Финансирование
Департамент здравоохранения г. Москвы , Российских фонд прямых инвестиций, ПАО «Сбербанк», ОК РУСАЛ.
Предшествующие данные
Мы провели поиск экспериментальных статей в PubMed, опубликованных до 25 января 2021 года, без ограничений по языку, с использованием терминов «SARS-CoV-2» или «COVID-19», «vaccine», «clinical trial» и «efficacy». Мы нашли три доступные прошедшие рецензию публикации, посвященные эффективности вакцин против SARS-CoV-2: AZD1222 (AstraZeneca/Оксфордский университет), вакцины, основанной для ChAdOx1, эффективность которой составила 70,4%, и двух вакцин на основе мРНК: BNT162b2 (Pfizer/BioNTech) с эффективностью 95% и mRNA-1273 (Moderna/NIAID) с эффективностью 94,1%. Мы ранее публиковали данные о безопасности и иммуногенности Гам-КОВИД-Вак в I-II фазах клинических испытаний.
Дополнительный вклад данного исследования
Мы публикуем промежуточные результаты оценки клинической эффективности основанной на векторах rAd26 и rAd5 вакцины Гам-КОВИД-Вак против COVID-19 в рандомизированном, двойном слепом плацебо-контролируемом многоцентровом исследовании III фазы в г. Москве, Россия, включающем 21 862 участников. Мы впервые описываем результаты по иммуногенности для данного исследования, в том числе титры рецептор-связывающих домен-специфичных IgG, титры нейтрализующих вирус антител и ответ IFN-γ. Вакцинация в гетерологичном режиме прайм-буст предоставляет стабильный гуморальный и клеточный иммунный ответ с 91,6% (95% ДИ 85,6–95,2) эффективностью против COVID-19. Температура хранения и распространения вакцины составляет –18° C, однако хранение при температуре 2–8 °C, предпочтительные условия для глобального распространения, также было одобрено Министерством здравоохранения Российской Федерации.
Общие выводы из всех имеющихся данных
Системный подход к борьбе с пандемией COVID-19 требует введения различных вакцин с разными механизмами действия, чтобы удовлетворить глобальные нужды здравоохранения с помощью рентабельных и подходящих для конкретного региона методов. Наша вакцина совместно с другими вакцинами против SARS-CoV-2 помогает разнообразить арсенал вакцин против этого вируса.
Введение
Пандемия COVID-19 привела к более чем 98 миллионам подтвержденных случаев заболевания и более чем к 2 миллионам смертей в мире (информация на дату публикации). Существуют предварительно лицензированные вакцины против COVID-19, а усилия специалистов сконцентрированы на разработке безопасных и эффективных вакцин для предотвращения COVID-19 prevention. Согласно черновику обзора кандидатных вакцин против COVID-19, составленному ВОЗ (1), 64 вакцины-кандидата находятся в стадии клинических испытаний (в том числе 13 в III фазе) и 173 — на стадии доклинического анализа. Вакцины-кандидаты, находящиеся на III фазе, включают разные платформы: векторые вакцины (Национальный исследовательский центр эпидемиологии и микробиологии имени почётного академика Н. Ф. Гамалеи [НИЦЭМ им. Н. Ф. Гамалеи; данное исследование], Оксфордский университет /AstraZeneca (2), CanSino Biological Inc/Пекинский институт биотехнологий и компания Janssen Pharmaceutical), мРНК-вакцины (Moderna/Национальный институт аллергии и инфекционных заболеваний (3) и BioNTech/Fosun Pharma/Pfizer (4), инактивированные вакцины (SinoVac, Уханьский институт биологических продуктов/Sinopharm, Пекинский институт биологических продуктов/Sinopharm и Bharat Biotech) и наночастинцы рекомбинантного белка с адъювантом (Novavax).
Безопасность аденовирусных векторных вакцин хорошо изучена, и лекарственные средства на основе аденовирусных векторов используются в клинической практике (5, 6, 7). Известно, что доставка антигенов аденовирусными векторами индуцирует как клеточный, так и гуморальный иммунитет после одного введения, что дает возможность использовать такие вакцины в качестве средства экстренной профилактики в условиях пандемии. Введение двух доз вакцины позволяет развить надёжный и долгосроыный иммунный ответ (8, 9). Эти характеристики делают рекомбинантные вакцины на основе дефектных по репликации аденовирусов подходящими кандидатами на роль целевых продуктов ВОЗ для долгосрочной защиты людей из групп высокого риска COVID-19 в условиях вспышки заболевания, поскольку такие вакцины стимулируют быстрое развитие защитного иммунитета. Аденовирусные вектора могут индуцировать иммунный ответ на компоненты самого вектора и снижать эффективность иммунного ответа на антиген, система гетерологичной вакцинации в режиме прайм-буст с двумя разными векторами позволяет минимизировать этот эффект (9, 10, 11).
Таким образом, наиболее эффективным подходом к созданию мощного и долгосрочного иммунного ответа, не зависящего от наличия предшествующего иммунного ответа на вектор, является подход гетерологичной вакцинации в режиме прайм-буст, который мы использовали при разработке вакцины для предотвращения COVID-19.
Гам-КОВИД-Вак — это комбинированная векторная вакцина, основанная на рекомбинантном аденовирусе (rAd) типа 26 (rAd26) и rAd типа 5 (rAd5); оба аденовируса несут полноразмерный ген гликопротеина S вируса SARS-CoV-2 (rAd26-S и rAd5-S). rAd26-S и rAd5-S вводятся внутримышечно раздельно с интервалом в 21 день. Клинические испытания I-II фазы были закончены в августе 2020 г. (12).
Результаты показали хорошую переносимость и высокую иммуногенность у здоровых участников. В результате вакцина-кандидат была предварительно одобрена в России в соответствии с национальным законодательством. Такая регистрация позволять использовать вакцину в группах высокого риска с усиленной фармакологической бдительностью во время проведения пострегистрационного исследования эффективности. В этой статье мы представляет предварительные данные по эффективности и безопасности III фазы многоцентрового исследования Гам-КОВИД-ВАК у взрослых с отдельным анализом возрастной группы старше 60 лет.
Методы
Дизайн исследования и подбор участников
Данное исследование — рандомизированное двойное слепое плацебо-контролируемое многоцентровое испытание III фазы для оценки эффективности, иммуногенности и безопасности комбинированной векторной вакцины Гам-КОВИД-Вак против индуцированного SARS-CoV-2 COVID-19 у взрослых, проведённое в 25 больницах и поликлиниках Москвы. Только аккредитованные Министерством здравоохранения Российской Федерации для проведения клинических исследований организации могли участвовать в исследовании. Протокол испытания был проверен и одобрен соответствующими компетентными организациями, включая Департамент государственного регулирования обращения лекарственных средств Министерства здравоохранения Российской Федерации (Разрешение № 450 от 25 августа 2020 г.), Московский городской независимый этический комитет и независимые местные этические комитеты в местах проведения испытания.
Стратегии привлечения участников для достижения высокого уровня участия в исследовании включали использование онлайн-платформы Официального сайта мэра Москвы и его колл-центров, программы работы с населением и усилия по привлечению в одобренных для проведения испытания клинических организациях. В исследование включали всех, кто подписал информированное согласие и прошел скрининг.
Критерии включения были следующими: возраст не менее 18 лет; отрицательные результаты анализов на наличие ВИЧ, гепатита B или C и сифилиса; отрицательные результаты анализов на наличие антител классов IgM и IgG против SARS-CoV-2, а также отрицательный результат ПЦР на SARS-CoV-2; отсутствие COVID-19 в анамнезе; отсутствие контактов с лицами с COVID-19 в течение предшествующих 14 дней; согласие на использование эффективных методов контрацепции; отрицательные тест на беременность по моче (для женщин детородного возраста); отрицательные результаты исследования на наркотики и алкоголь в момент скрининга; отсутствие индуцированных вакциной реакций в анамнезе; отсутствие острых инфекций или респираторных заболеваний в течение 14 дней, предшествующих включению.
Критериями исключения из исследования была любая вакцинация в течение 30 дней, предшествовавших включению; иммуносупрессия в течение 3 месяцев, предшествовавших включению; беременность или кормление грудью; острый коронарный синдром или инсульт в течение года, предшествовавшего моменту включения; туберкулёз или хронические системные инфекции; аллергия или гиперчувствительность к препарату или его компонентам; новообразования; донорство крови в течение двух месяцев, предшествовавших включению; спленэктомия; нейтропения; агранулоцитоз; существенная потеря крови, тяжелая анемия или иммунодефицит в течение предыдущих 6 месяцев; активная форма заболевания, вызванного ВИЧ, сифилиса, гепатита B или C; анорексия или недостаток белка; наличие крупных татуировок в месте инъекции; алкоголизм или зависимость от наркотиков в анамнезе; участие в любом другом клиническом испытании; сотрудники центра исследований или другие сотрудники, непосредственно участвовавшие в проведении исследования, и члены их семей; а также любые другие обстоятельства, которые проводящий исследование терапевт счел проблемными. Все участники предоставили подписанное информированное согласие на включение в базу данных для участия в исследовании.
Рандомизация и ослепление
Участники исследования были разделены на пять возрастных групп (18–30, 31–40, 41–50, 51–60 и >60 лет) и были распределены на две группы с использованием стратифицированной (размер блока 4) интерактивной системы рандомизации с доступом через веб-портал в соотношении 3:1 группы вакцины к контрольной группе. Участники исследования получили уникальные рандомизационные номера, остававшиеся неизменными до окончания исследования. Статистик создал последовательность, в соответствии с которой были промаркированы препараты. Препарат и плацебо были неразличимы внешне (по упаковке, маркировке и содержанию). Исследователи, участники и все сотрудники, участвовавшие в организации исследования, не обладали информацией о распределении по группам.
Процедуры
Все добровольцы, согласившиеся участвовать в исследовании, совершили скрининговый визит для медицинского осмотра, проверки показателей жизненно важных функций (артериальное давление, частоты сердечных сокращений и температуры тела), анализа крови на инфекции (ВИЧ, гепатит B и C, сифилис) и сбор исходных характеристик иммуногенности. Для всех участников было проведено исследование мочи на алкоголь и наркотики; для женщин был также проведён тест на беременность. В рамках скрининга в центральной лаборатории в Москве были также методом ПЦР на SARS-CoV-2 исследованы мазки для исключения участников с COVID-19. При скрининге в индивидуальную регистрационную карту каждого участника была внесена информация о сопутствующих заболеваниях и группе риска инфекции SARS-CoV-2. Группа высокого риска включала тех участников, работа которых предполагает взаимодействие с пациентами с подтвержденным диагнозом COVID-19; группа среднего риска — тех, кто в связи со своей работой контактирует с большим количеством людей (например, врачи общей практики, социальные работники и сотрудники магазинов); группа общего риска — тех, кто не обладает дополнительными рисками в связи со своей работой. Предполагаемая длительность участия в испытании составляла 180 дней после первой дозы вакцины или плацебо. В ходе испытания были запланированы один скрининговый визит и пять посещений центра проведения вакцинирования.
Вакцина состоит из двух векторных компонентов, rAd26-S и rAd5-S. Полная доза вакцины содержит 1011 на дозу каждого из рекомбинантных аденовирусов; 0,5 мл на дозу для внутримышечный инъекции. Плацебо состоит из компонентов вакцинного буфера, однако не содержит рекомбинантных аденовирусов; объем соответствует объему вакцины. Вакцина и плацебо разработаны, выпущены и хранились в НИЦЭМ им. Н. Ф. Гамалеи в соответствии с надлежащей производственной практикой. Вакцина и плацебо использовались в жидкой (замороженной) форме. Состав вакцины и плацебо описан в приложении (с. 1). Вакцину (первая доза — rAd26, вторая — rAd5) или плацебо вводили внутримышечно в дельтовидную мышцу с 21-дневным интервалом между дозами.
Последующие посещения для наблюдения были запланированы на 28-й (±2 дня), 42-й (±2 дня) и 180-й день (±14 дней). При посещениях для наблюдения у всех участников оценивали показатели жизненно важных функций и записывали изменения в состоянии и самочувствии участников по сравнению с предыдущим посещением. В день введения второй дозы (21-й день) проводили ПЦР-тест в комбинации с клиническим обследованием для диагностики симптоматического и несимптоматического COVID-19. При наличии клинических признаков респираторной инфекции и положительного ПЦР-теста участник не получал вторую дозу и обращался к врачам для лечения COVID-19. Участники без признаков респираторной инфекции были вакцинированы для получения результатов ПЦР. В случае положительного результата ПЦР участники считались бессимптомными носителями COVID-19 и не учитывались как случае COVID-19 в анализе эффективности, согласно протоколу. В течение исследования, кроме дня скринингового исследования и дня введения второй дозы, дополнительных ПЦР-исследований проведено не было за исключением случаев, когда участники сообщали о появлении у них симптомов COVID-19.
Спонсор предоставил возможность дополнительных наблюдений с помощью телемедицинского обслуживания. Участникам исследования было рекомендовано использовать эти незапланированные телемедицинские консультации для для жалоб или вопросах о процедурах исследования. Все участники получили контакты команды исследования в момент подписания информированного согласия и рекомендацию связываться с исследователями по необходимости, однако в первую очередь при появлении любых признаков или симптомов, которые могли бы свидетельствовать о нежелательных явлениях. Всем участникам была также предоставлена возможность вести электронные дневники, устанавливаемые на смартфоны, для отслеживания состояния их здоровья. Информацию от участников, которые предпочли не использовать электронные дневники, получали в ходе телеконсультаций. Информацию, собранную в ходе телемедицинских консультаций, исследователи на местах добавляли непосредственно в медицинскую карту участника.
В г. Москве работает общегородская электронная медкарта и единая медицинская информационно-аналитическая система города Москвы (ЕМИАС). ЕМИАС — это контролируемая электронная медкарта, которую используют все организации здравоохранения Москвы для оказания медицинской помощи жителям г. Москвы. Единые электронные медкарты участников клинического испытания были обновлены (указано их участие в испытании) и были использованы в качестве источника электронных данных и проверки исходных данных контрактной наблюдающей исследовательской организацией. В дополнение к определёнными протоколом посещениями и телекоммуникациями, руководители и команда исследования могли наблюдали статус пациента через ЕМИАС, в том числе возможные случаи госпитализации и использование амбулаторных услуг. Электронные дневники участников, которые согласились их вести, были также интегрированы в электронную медкарту ЕМИАС. В случае тех участников, которые не согласились вести электронные дневники, информация о статусе участника была собрана сотрудниками центров и введена в электронную медкарту. Врачи-исследователи вели все нежелательные явления вплоть до разрешения; каждый случай был рассмотрен советом по безопасности данных и наблюдению и утверждён наблюдателями испытания. В случае подозрения на COVID-19 его проверяли в соответствии с диагностическим протоколом COVID-19, включающим ПЦР-тестирование в центральной лаборатории Москвы. Тяжесть заболевания устанавливали исследователи на местах после подтверждения диагноза COVID-19. Описание критериев оценки тяжести COVID-19 доступно в приложении (с. 2).
Исследование было организовано и находилось под наблюдением московского представительства контрактной исследовательской организации Crocus Medical (головной офис находится в Нидерландах). Управление данными осуществляется через систему DM 365 MainEDC (разработана Data Management 365), мощную облачную платформу, интегрирующую функции сбора данных, передовых технологий рандомизации, полного контроля над поставкой и распределением препаратов и электронных дневников пациентов (захват электронных данных, интерактивная система с доступом через веб-портал, снабжение препаратами и предоставленная пациентами электронная информация об исходах). Система соответствует всем применимым международным документам, включая часть 11 раздела 21 Свода федеральных нормативных актов США, GMP, GAMP 5, Акт о мобильности и подотчетности медицинского страхования (HIPAA) и общему регламенту по защите данных (GDPR). Система позволяет собирать и валидировать клинически данные в клинических испытаниях с большой нагрузкой и поддерживает центральный мониторинг и мониторинг, основанный на анализе рисков, кодирование с использованием Медицинского словаря для регуляторной деятельности (MedDRA), WHODrug и базы наименований и кодов идентификаторов логического наблюдения (LOINC), а также мгновенное соотнесение экспортированных данных со стандартом модели табулирования Консорциума стандартов обмена клиническими данными.
Взятие крови производили в день вакцинации непосредственно перед введением препарата. Взятие крови для оценки параметров иммуногенности проводили только в некоторых центрах исследования, выбранных на основе логистики для доставки биоматериалов в центральную лабораторию, где проводили первичную обработку крови (сбор сыворотки, разделение на партии и заморозку). Взятие образцов крови у вплоть до 9520 участников исследования запланировано перед завершением клинического испытания.
Иммуногенность анализировали согласно опубликованному ранее методу (12). Его суть состоит в анализе антиген-специфичного гуморального иммунного ответа в день первой вакцинации и на 42-й день. Титр антител, специфичных к гликопротеину, в сыворотке крови, выявляли с помощью иммуноферментного анализа (ИФА). Для тестирования на IgG против SARS-CoV-2 мы использовали метод ИФА, разработанный в НИЦЭМ им. Н. Ф. Гамалеи и зарегистрированный для клинического использования в России (P3H 2020/10393 2020-05-18). ИФА позволяет измерять IgG, специфичные к рецептор-связывающему домену (RBD) S-гликопротеина SARS-CoV-2. Титр нейтрализующих антител измеряли в день первой вакцинации и на 42-й день методом микронейтрализации с использованием SARS-CoV-2 (hCoV-19/Russia/Moscow_PMVL-1/2020) в 96-луночных планшетах с полуинфицирующей дозой культуры ткани (TCID50) 100. Уровень сероконверсии рассчитывали на четырёхкратное повышение титра на 42-й день по сравнению с днём перед первой вакцинацией. Клеточный иммунный ответ измеряли в день первой вакцинации и на 28-й день с помощью изменения секреции IFN-γ при повторной стимуляции антигеном в культуре мононуклеарных клеток периферической крови.
Исходы
Основным исходом являлась доля участников с подтвержденным ПЦР диагнозом COVID-19 начиная с 21-го дня после получения первой дозы. Вторичными исходами были: тяжесть COVID-19; изменение уровня антител против гликопротеина S SARS-CoV-2; доля участников с антителами против N-белка SARS-CoV-2; изменение (увеличение) титра нейтрализующих SARS-CoV-2 антител; изменение уровня антиген-специфичного клеточного иммунного ответа (усиление опосредованного иммунными клетками ответа на антиген); и наличие и тяжесть нежелательных явлений. Серьезные нежелательные явления были диагностированы на основе случаев, когда была необходима госпитализация. В этом отчёте представлены предварительные данные измерения основного исхода, частоты и тяжести возникновения нежелательных явлений, иммуногенности и безопасности.
Статистический анализ
В этом промежуточном анализе мы представляем данные об эффективности на этапе подтверждения 78 случаев COVID-19 у участников исследования после введения второй дозы в соответствии с протоколом.
В этом исследовании основным исходом являлась доля участников без подтвержденного лабораторными исследованиями COVID-19 в течение исследования. Частота COVID-19 в общей популяции и, соответственно, ожидаемая частота в группе плацебо, составляет 20 на 1000 человек, или 2,0%. Цель этого исследования — показать, что доля участников с COVID-19 будет хотя бы на треть ниже в экспериментальной группе, чем в контрольной (отношение шансов [ОШ] для нулевой гипотезы 0,67), т.е. верхний порог для 95% ДИ для ОШ не должен превысить 0,67. Ожидаемое значение эффекта составляет около 0,500 (ОШ для альтернативной гипотезы 0,500). С запланированным числом 40 000 участников и рандомизацией 3:1 (вакцина: плацебо) мощность исследования составит 85% с односторонним уровнем статистической значимости 0,025.
Протокол исследования исходно не указывал целевое число событий в испытании. Тем не менее, в связи с увеличением частоты заболеваемости COVID-19 в России в протокол клинического испытания 5 ноября 2020 были внесены изменения, включающие промежуточный анализ для предварительной оценки эффективности вакцины и установления этической целесообразности дальнейшего включения группы плацебо в испытание в контексте развития пандемии в случае эффективности вакцины. Три промежуточных анализа были проведены, когда в обеих группах в сумме были зарегистрированы 20, 39 и 78 документированных случаев COVID-19. Наша исходная консервативная оценка эффективности составляла 50%. Если эффективность составляет хотя бы 70%, статистически значимые различия между группами можно зарегистрировать при наличии хотя бы 20 событий в двух группах. Если эффективность составляет не менее 70%, число необходимых событий составляет 39. 60% эффективность даст статистически значимую разницу при 78 зарегистрированных случаях.
ОШ и 95% ДИ рассчитывали согласно описанным ранее методам (13).
Основной исход рассчитывали по следующей формуле:
Эффективность вакцины (%)=(1 – ОШ) × 100, где ОШ = OR=(a/b) / (c/d) =(a×d) / (b×c), где a — число вакцинированных участников с COVID-19, b — число вакцинированных участников без COVID-19, c — число невакцинированных участников с COVID-19, а d — число невакцинированных участников без COVID-19.
ОШ и 95% ДИ были рассчитаны по методу Баптисты — Пайка, p-значения были получены с помощью критерия χ2 или точного критерия Фишера (если ожидаемая частота хотя бы в одной из ячеек составляла менее 5). Кумулятивная частота показана с помощью метода Каплана-Мейера.
При анализе безопасности нежелательные события были закодированы с помощью MedDRA версии 23.0. Нежелательные события показаны по группе, системе органов, классу и предпочтительному термину. Нормальность распределения данных при анализе количественных данных в анализе иммуногенности оценивали с помощью критерия д’Агостино — Пирсона. При анализе иммуногенности (анализ параметрических данных) в случаях, когда сравнивали две группы данных, использовали U-критерий Манна — Уитни (например, сравнение группы вакцины и группы плацебо или мужчин и женщин) для непарных образцов и критерий знаковых рангов Вилкоксона для парных образцов (например, данные по клеточному ответу в дни до и после вакцинации). При сравнении нескольких групп (например, возрастных) использовали критерий Краскела — Уоллиса. Для сравнения показателей частоты между группами использовали критерий χ2 и при необходимости точный критерий Фишера (если ожидаемая частота в какой-либо ячейке составляла меньше 5).
В анализ основного исхода включали всех участников, которые получили хотя бы две дозы к моменту закрытия базы данных и следовали протоколу без нарушений. Анализ серьёзных нежелательных эффектов включал всех участников, которые получили хотя бы одну дозу к моменту закрытия базы данных и следовали протоколу без нарушений. В анализ безопасности (в том числе редких нежелательных явлений) включали всех участников, которые получили две дозы и для которых все доступные данные были проверены на момент закрытия базы данных. Статистический анализ проведен с помощью программ Stata версии 14 и GraphPad Prism версии 9.0. Клиническое исследование зарегистрировано в системе ClinicalTrials.gov (NCT04530396).
Роль источника финансирования
Источник финансирования не оказывал влияния на дизайн исследования, сбор, анализ и интерпретацию данных или написание отчёта.
Результаты
В промежутке с 7 сентября по 24 ноября 2020 года 21 977 взрослых участников подходили по критериям включения и были случайным образом распределены в группу плацебо (n=5476) или вакцины (n=16 501; рис. 1).
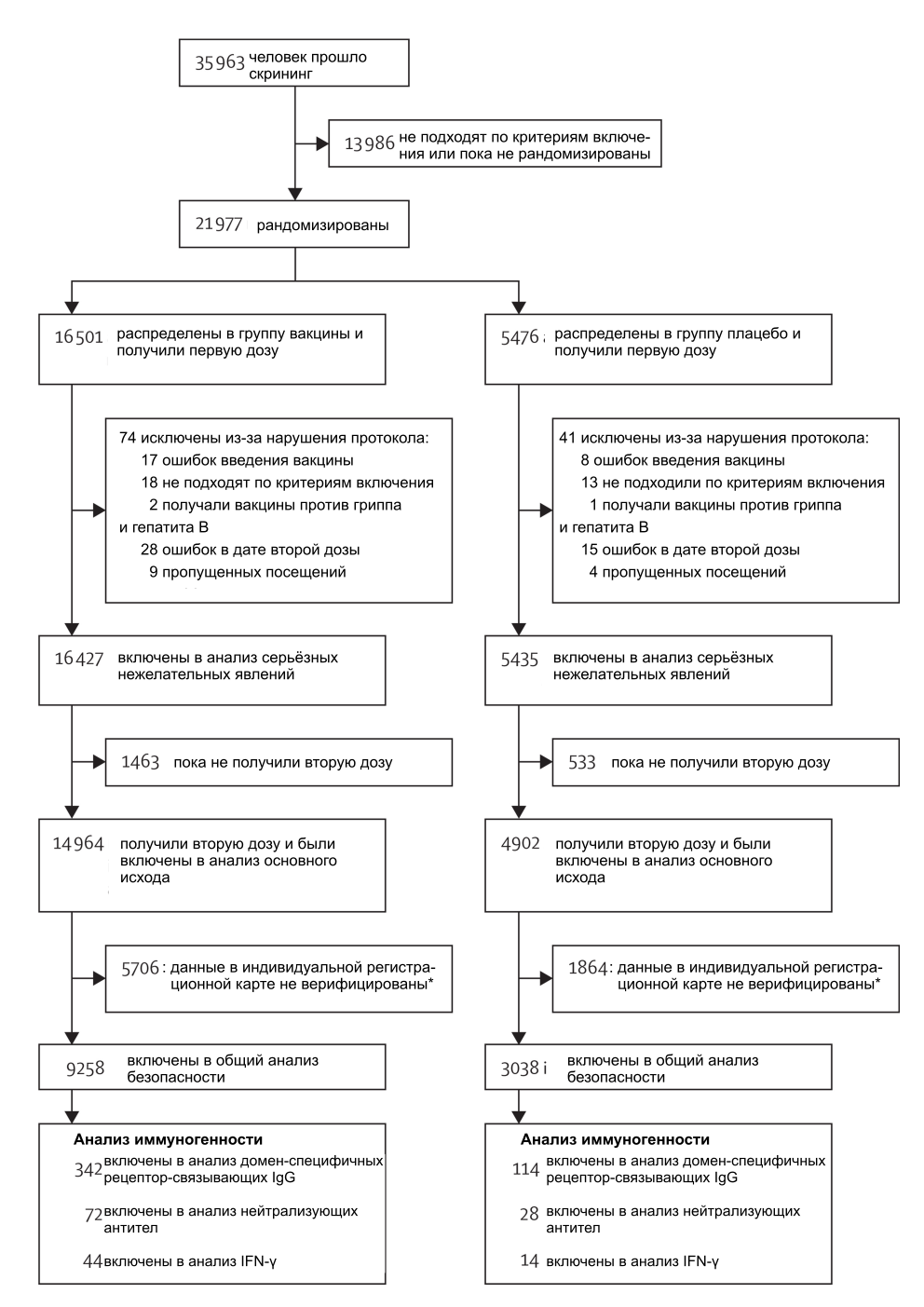
*На момент закрытия базы данных данные о нежелательных явлениях в регистрационных картах этих участников не были верифицированы; процедура верификации данных может производиться с небольшой задержкой; следовательно, участники, данные которых не были верифицированы, не были включены в этот анализ.
Первое закрытие базы данных было осуществлено 18 ноября 2020 г. с достижением 20 случаев COVID-19. Промежуточный анализ безопасности (анализ редких нежелательных явлений) был проведён с данными, полученными к первому закрытию базы данных. В связи с увеличением количества случаев в Москве в течение ноября второе закрытие базы данных было осуществлено 24 ноября 2020 г. с достижением 78 случаев COVID-19. Представленные данные для промежуточного анализа эффективности и серьезным нежелательным явлениям получены в момент второго закрытия базы данных.
74 участника из группы вакцины и 41 участник из группы плацебо были исключены из анализа (рис. 1). Этот предварительный анализ включал 16 427 участников в группе вакцины и 5435 в группе плацебо, которые получили хотя бы одну дозу и продолжили участие в исследовании. 14 964 участников в группе вакцины и 4902 в группе плацебо получили две дозы к моменту закрытия базы данных (24 ноября 2020 г.) и были включены в анализ основного исхода (таблица 1). Медианное время между получением участником первой дозы до закрытия базы данных составило 48 дней (интерквартильный размах 39–58). Средний возраст участников, получивших две дозы, составил 45,3 лет (стандартное отклонение 12,0) в группе вакцины и 45,3 лет (стандартное отклонение 11,9) в группе плацебо; распределение по полу (p=0,619), частота встречаемости сопутствующих заболеваний (p=0,420) и риск инфицирования (p=0,851) в двух группах были схожи (таблица 1).
Таблица 1. Исходные характеристики участников, получивших две дозы назначенного препарата и включенных в анализ основного исхода

Данные представлены в виде % и средних (стандартное отклонение).
* Включает афроамериканцев, аборигенов Гавайских или других тихоокеанских островов, а также участников с неизвестной расовой принадлежностью.
† Знаменатель показывает число участников, для которых были доступны данные.
‡ Группа высокого риска включала тех участников, работа которых предполагает взаимодействие с пациентами с подтвержденным диагнозом COVID-19; группа среднего риска — тех, кто в связи со своей работой контактирует с большим количеством людей (например, врачи общей практики, социальные работники и сотрудники магазинов); группа общего риска — тех, кто не обладает дополнительными рисками в связи со своей работой.
От 21-го дня после первой дозы вакцины (день введения второй дозы) в группе вакцины были подтверждены 16 случаев COVID-19 (0,1% среди 14 964 участников), в то время как в группе плацебо были подтверждены 62 случая (1,3% из 4902 участников); эффективность вакцины составила 91,6% (95% ДИ 85,6–95,2; таблица 2). Наблюдаемая эффективность вакцины превышала 87% во всех подгруппах по возрасту и полу. Следует отметить, что эффективность вакцины составила 91,8% (67,1–98,3) у участников старше 60 лет. Не было ни одного случая в группе вакцины против 20 случаев в группе плацебо умеренно тяжелого или тяжелого течения COVID-19, подтвержденного хотя бы через 21 день после введения первой дозы; следовательно, эффективность вакцины против умеренно тяжелого или тяжелого COVID-19 составляет 100% (94,4–100,0). В промежутке с 15 до 21 дней после введения первой дозы эффективность составила 73,6% (p=0,048), а затем начиная с 21-го дня — 100% (p<0,0001; с. 11 в приложении).
Таблица 2. Промежуточные результаты анализа эффективности вакцины

Данные представлены как n/N (%), если не указано иное.
* Включает участников, получивших две дозы.
† Включает участников, получивших хотя бы одну дозу.
97 подтвержденных случаев COVID-19 (63 в группе вакцины и 34 в группе плацебо) не отражены в анализе первичного исхода, поскольку они произошли менее чем через 21 день после первой дозы (т.е. до введения второй дозы; таблица 2, рисунок 2). Согласно оценке, эффективность вакцин против подтвержденного COVID-19 в любое время после первой дозы составила 73,1% (95% ДИ 63,7–80,1). Следует отметить, что в группе вакцины большинство случаев COVID-19 произошли до второй дозы. Частоты выявления заболевания были схожими в группах вакцины и плацебо вплоть до 16–18 дней после первой дозы, после чего раннее появление защиты привело к гораздо более медленному накоплению новых случаев в группе вакцины, чем в группе плацебо (рисунок 2).

Рисунок 2. Кумулятивная кривая Каплана — Мейера для накопления случаев симптоматического ПЦР-положительного COVID-19 после введения первой дозы среди участников исследования, которые получили хотя бы одну дозу вакцины или плацебо.
Промежуточный анализ иммуногенности включал образцы, транспортированные из центральной лаборатории, которые были собраны до 30 ноября 2020 г., и показал, что вакцина индуцирует иммунный ответ у участников. Перед первой вакцинацией ни у кого из участников в плазме крови не было детектировано специфичных к RBD антител (проанализированы 456 участников) или нейтрализующих вирус антител (проанализированы 100 участников). При анализе гуморального иммунного ответа образцы сыворотки крови 456 участников (342 из группы вакцины и 114 из группы плацебо) изучили на предмет наличия антител, специфичных к рецептор-связывающему домену S-гликопротеина SARS-CoV-2 через 42 дня после начала вакцинации (рисунок 3A). В группе вакцины специфичные к RBD IgG были зарегистрированы у 336 (98%) из 342 образцов с геометрическим средним титром (GMT), равным 8996 (95% ДИ 7610–10 635) и скоростью сероконверсии 98,25%. В группе плацебо специфичные к RBD IgG были зарегистрированы в 17 (15%) из 114 образцов со значением GMT 30,55 (20,18–46,26) и скоростью сероконверсии 14,91% (p<0,0001 при сравнении с группой вакцины). При сравнении уровней специфичных к RBD антител между возрастными группами мы обратили внимание на то, что у возрастной группы 18–30 (данные для женщин и мужчин оценивали совместно) значение GMT было существенно выше, чем у остальных возрастных групп (p=0,0065). Разницы между другими возрастными группами не было (p=0,343). Уровень антител также статистически значимо не различался у мужчин (n=179) и женщин (n=159; p=0,258). Описательные статистики по возрастным и половым группам представлены в приложении (с. 3).
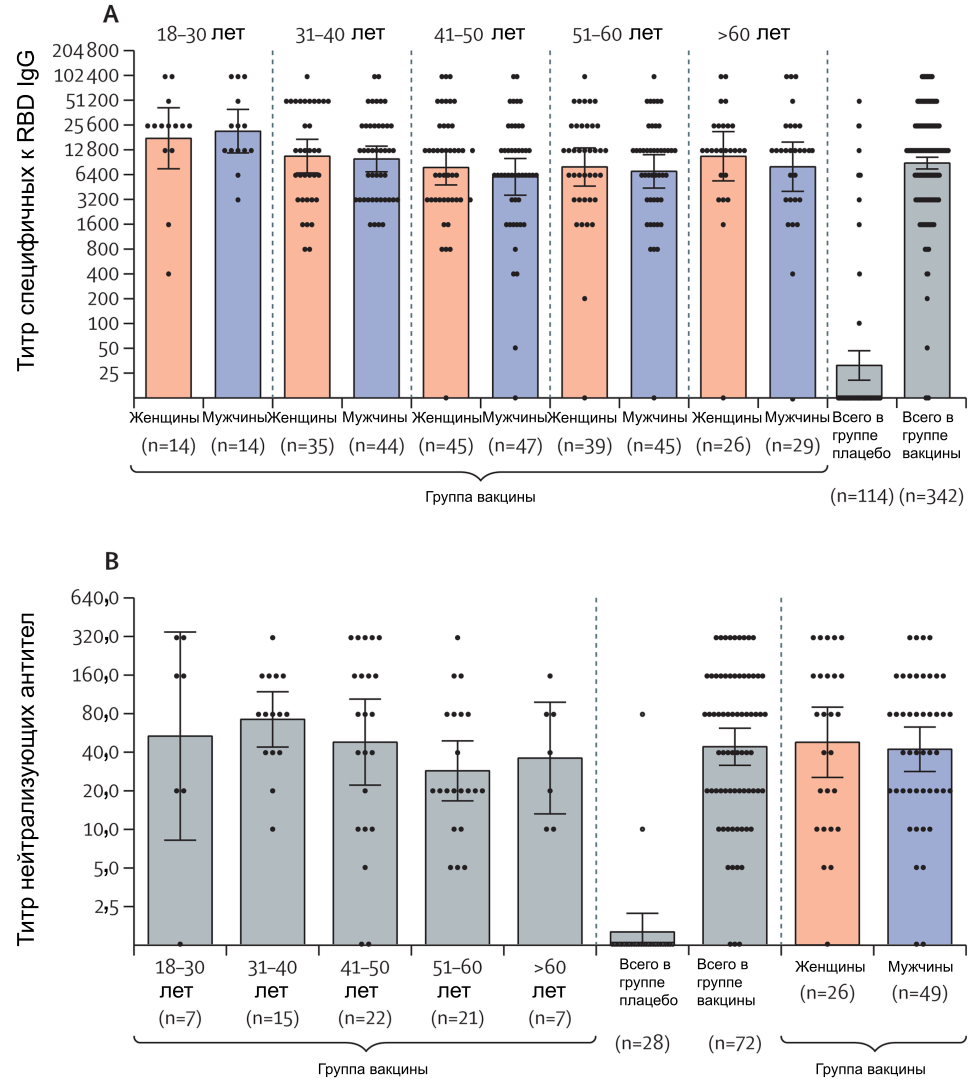
Рисунок 3. Гуморальный иммунный ответ. (A) Уровень антител, специфичных к рецептор-связывающему домену, измеренный с помощью ИФА, у пациентов, которым была введена вакцина (по возрастной группе и общая оценка по группе), и пациентов, которым было введено плацебо (общая оценка по группе). (B) Уровень нейтрализующих антител на 42-й день по данным анализа нейтрализации (TCID50 = 100) у участников, которым была введена вакцина или плацебо. Данные разделены по возрастным группам и полу. Также представлены общие данные по группам вакцины и плацебо. Точки соответствуют индивидуальным измерениям, столбики — геометрическим средним, а планки погрешностей отмечают 95% ДИ. TCID50=полуинфицирующая доза культуры ткани.
Для оценки индукции гуморального иммунного ответа образцы плазмы крови 100 участников проанализировали на предмет наличия нейтрализующих антител на 42-й день после первой вакцинации (рисунок 3B); в группе вакцины GMT нейтрализующих антител составил 44,5 (95% ДИ 31,8–62,2), а уровень сероконверсии — 95,83%. GMT в группе плацебо составил 1,6 (1,12–2,19), а уровень сероконверсии составил 7,14%, что значительно ниже, чем в группе вакцины (p<0,0001). Уровень нейтрализующих антител был сходным у разных возрастных групп (p=0,222), а также у мужчин и женщин (p=0,639). Описательные статистики по возрастным группам и полу представлены в приложении (с. 3). Клеточный иммунный ответ у участников характеризовали по секреции IFN-γ мононуклеарными клетками периферической крови при повторной стимуляции S-гликопротеином SARS-CoV-2 в культуре. Для оценки клеточного иммунного ответа проанализировали образцы плазмы крови 58 участников (44 из группы вакцины и 14 из группы плацебо). К 28-му дню после первой вакцинации у всех участников из группы вакцины наблюдали существенно более высокий уровень секреции IFN-γ после повторной стимуляции антигеном (медиана 32,77 пг/мл [интерквартильный размах 13,94–50,76]) по сравнению с днём введения первой дозы (рисунок 4). Описательные статистики иммунного ответа, связанного с IFN-γ, представлены в приложении (с. 4).
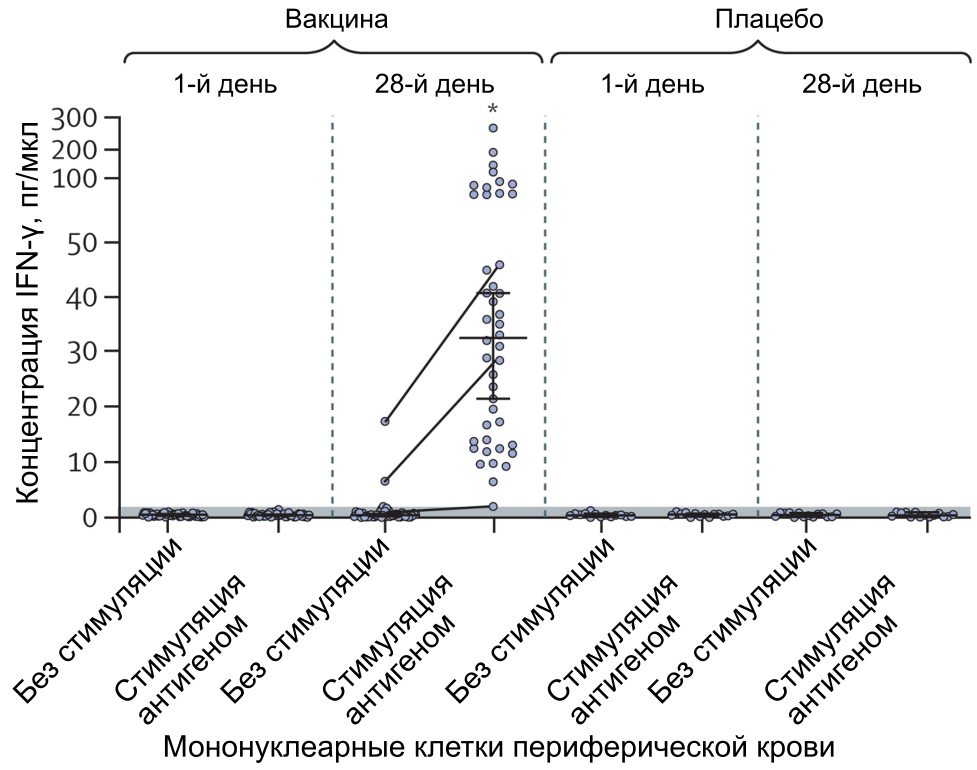
Рисунок 4. Ответ IFN-γ на S-гликопротеин SARS-CoV-2 в мононуклеарных клетках периферической крови участников, получивших две дозы вакцины (n=44) или плацебо (n=14). Точки соответствуют индивидуальным измерением интактных клеток (без стимуляции) и клеток, стимулированных S-гликопротеином SARS-CoV-2 (стимуляция антигеном). Горизонтальные линии показывают медианные значения; верхняя и нижняя планки — 95% ДИ. Порог обнаружения (<2 пг/мл) обозначен серой областью. Серые линии, соединяющие точки между клетками без стимуляции и после стимуляции антигеном показывают изменения в ответе IFN-γ у некоторых участников. *p<0,0001 для сравнения стимулированных антигеном клеток на 28-й день и на 1-й день в группе вакцины.
В анализ общей безопасности и редких нежелательных явлений были включены 12 296 участников, которые получили обе дозы до закрытия базы данных 18 ноября 2020. Наиболее частые нежелательные явления включали гриппоподобное заболевание, реакции на месте инъекции, головную боль и общую слабость. Большинство зарегистрированных нежелательных явлений (7485 [94,0%] из 7966) относились к первой степени тяжести; 451 (5,66%) — ко второй, а 30 (0,38%) — к третьей. В исследовании были зафиксированы 122 редких нежелательных явления (91 в группе вакцины и 31 в группе плацебо; приложение, с. 8–9).
В анализ серьезных нежелательных явлений были включены 21 862 участников, которые получили хотя бы одну дозу (19 866 из них получили две дозы) на момент закрытия базы данных 24 ноября 2020. 70 эпизодов серьезных нежелательных явлений были зарегистрированы у 68 участников исследования: 45 (0,3%) из 16 427 участников в группе вакцины и 23 (0,4%) из 5435 участников в группе плацебо (приложение, с. 5–7). Ни один случай не рассматривается как связанный с вакцинацией, что было подтверждено независимой наблюдательной комиссией.
Небольшое количество серьёзных нежелательных явлений позволяет проанализировать и верифицировать соответствующие данные вплоть до второго закрытия базы; тем не менее, полные данные о нежелательных явлениях, которые пока не были обработаны, будут представлены в публикациях в дальнейшем во избежание разночтений с окончательным отчетом после обработки всех данных.
В течение исследования были зафиксированы 4 смерти (3 [<0,1%] из 16 427 в группе вакцины и 1 [<0,1%] из 5435 участников в группе плацебо). Смертей, связанных с вакциной, не зарегистрирована. Одна смерть в группе вакцины были связана с переломом грудного позвонка; ещё две были связаны с COVID-19 (один пациент с тяжелыми нарушениями работы сердечно-сосудистой системы, симптомы проявились на 4-й день после введения первой дозы, и один пациент с коморбидной патологией эндокринной системы, симптомы проявились на 5-й день после введения первой дозы; приложение, с. 12). На основе известной информации об инкубационном периоде заболевания можно предположить, что оба участника, вероятно, были инфицированы COVID-19 еще до включения в исследование, несмотря на отрицательный результат ПЦР. Смерть в группе плацебо была связана с геморрагическим инсультом.
В исследование были включены 2144 участника старше 60 лет (1611 в группе вакцины и 533 в группе плацебо). Средний возраст в этой группе составлял 65,7 лет (стандартное отклонение 4,5) в группе вакцины и 65,3 лет (4,3) в группе плацебо (приложение, с. 10). Максимальный возраст участников составлял 87 лет в группе вакцины и 84 года в группе плацебо. Соотношение участников в полу (p=0,378), частота сопутствующих заболеваний (p=0,774) и риск инфекции (p=0,090) были сходными в группах вакцины или плацебо. Переносимость вакцины у этих участников была хорошей. 1369 участников старше 60 лет (тех, кто получил две дозы и для кого данные в регистрационной карте были верифицированы к моменту закрытия базы данных [18 ноября 2020]) были включены в анализ безопасности. Наиболее частыми нежелательными явлениями были гриппоподобные заболевания у 156 (15,2%) и местная реакция 56 (5,4%) у 1029 участников в группе вакцины и 30 (8,8%) и 4 (1,2%) из 340 участников в группе плацебо (приложение, с. 11). Были зарегистрированы 3 эпизода нежелательных явлений третьей степени или тяжелее, которые не следует считать связанными с вакцинацией: обострение мочекаменной болезни и острый синусит в группе вакцины и гриппоподобное заболевание в группе плацебо. Все эти нежелательные явления были разрешены. Среди участников старше 60 лет в группе вакцины были зарегистрированы три серьезных нежелательных явления: почечная колика и тромбоз глубоких вен (оба события были ассоциированы с предсуществующими коморбидными патологиями) и абсцесс конечности (в связи с травмой и последующей инфекцией раневой поверхности мягких тканей пальца). Никакой связи между серьезными нежелательными явлениями и введением вакцины выявлено не было.
Обсуждение
Наши промежуточные результаты III фазы испытаний Гам-КОВИД-Вак показывают, что вакцина на 91,6% (95% ДИ 85,6–95,2) эффективна против COVID-19 (с 21-го дня после первой дозы и дня введения второй дозы). Наши результаты также показали, что вакцина на 100% (95% ДИ 94,4–100) эффективна против тяжёлого COVID-19, хотя этот исход был вторичным, и поэтому результаты являются промежуточными. Вакцина хорошо переносилась: 45 (0,3%) из 16 427 участников сообщили о серьезных нежелательных явлениях, все из которых следует рассматривать как не имеющие отношения к вакцине. В соответствии с дизайном исследования, начальной точкой для отсчёта случаев COVID-19 для оценки эффективности вакцины был 21 день после первой дозы (день введения второй дозы). Хотя дизайн данного исследования не предполагал оценку эффективности введения одной дозы, выбор ранней стартовой точки позволяет нам наблюдать возможный частичный защитный эффект введения одной дозы. Кумулятивные кривые заболеваемости COVID-19 в группах плацебо и вакцины начинают различаться на 16-18-й день после первой иммунизации, показывая раннее наступление частичного защитного эффекта после введения одной дозы; тем не менее, дизайн исследования не позволяет нам делать выводы из этих наблюдений.
Вакцина индуцировала надёжный гуморальный (n=342) и клеточный (n=44) иммунный ответ во всех возрастных группах. Следует отметить наличие нескольких участников, не давших клинического ответа, в группе вакцины (6 из 342); возможными причинами могут быть «старение» иммунной системы у людей старшего возраста, индивидуальные характеристики формирования иммунного ответа или сопутствующие иммунологические заболевания.
17 (15%) из 114 участников в группе плацебо обладали специфичными к RBD антителами на 42-й день, что, вероятно, связано с бессимптомным COVID-19; тем не менее, у всех этих участников ПЦР-тест на SARS-CoV-2 был отрицательным, и никто из них не сообщал о симптомах респираторного заболевания в электронном дневнике или при опросе при телемедицинской консультации.
В связи с большой важность защиты людей старшего возраста как группы риска мы оценили способность вакцины индуцировать иммунный ответ и защищать от COVID-19 у лиц старше 60 лет. Наши данные показывают, что двухкомпонентная вакцина Гам-КОВИД-Вак способна индуцировать нейтрализующий вирус гуморальный иммунный ответ у участников старше 60 лет. Более того, эффективность вакцины в этой группе участников существенно не отличалась от таковой в возрастной группе 18-60 лет.
Ограничения промежуточного анализа эффективности включают небольшой размер выборки внутри возрастных групп. Дальнейший сбор данных позволит уточнить эффективность в разных возрастных группах. Кроме того, случаи COVID-19 были зарегистрированы на основании сообщений самих пациентов с последующим ПЦР-тестом; следовательно, в анализ эффективности были включены только симптоматические случаи COVID-19.
Исходно мы разработали две формы вакцины: жидкую (хранится при –18 °C) и сухую и сублимированную (хранится при 2–8 °C). В этом исследовании мы изучали жидкую форму, для которой необходимо хранение при –18 °C. Хранение при температуре 2–8 °C, предпочтительные условия для глобального распространения, также было одобрено Министерством здравоохранения Российской Федерации.
Мы ранее сообщали о локальных и системных нежелательных реакциях после введения Гам-КОВИД-Вак на небольшой выборке участников (12). В этом промежуточном отчете мы проанализировали данные о серьезных нежелательных реакциях у более чем 21 000 участников (более чем 16 000 из которых получили вакцину).
У 68 участников из двух групп зарегистрировано 70 эпизодов серьезных нежелательных реакций; ни один случай не следует рассматривать как связанный с вакцинацией. В течение исследования были зафиксированы четыре смерти: три в группе вакцины и одна в группе плацебо — ни одну из которых не следует считать связанной с вакцинацией, было подтверждено независимой наблюдательной комиссией. Никто из этих участников не сообщал о нежелательных эффектах после вакцинации.
Две связанные с COVID-19 смерти были обусловлены усугублением ранее присутствовавших сердечно-сосудистых и эндокринологических проблем. На основе данных о длине инкубационного периода, предоставленных ВОЗ и Центром по контролю и профилактике заболеваний (CDC) (14, 15) можно предположить, что эти два участника уже были инфицированы SARS-CoV-2 на момент рандомизации и вакцинации. На основе рекомендаций ВОЗ и CDC и обзора клинических данных наблюдательная комиссия подтвердила, что эти участники уже были инфицированы, и болезнь прогрессировала до формирования иммунитета под действием вакцины. Подробное описание состояния участников с COVID-19 доступно в приложении (с. 12). Среди 7 участников в группе плацебо, у которых был подтвержден COVID-19 в течение 7 дней после введения первой дозы, не было участников с коморбидными патологиями, в противоположность группе вакцины, в которой были 3 участника с коморбидными патологиями среди 25, у которых был подтвержден COVID-19 в течение первых 7 дней.
Таким образом, при рассмотрении двух случае связанных с COVID-19 смертей следует принимать во внимание ряд принципиально важных моментов. Во-первых, несмотря на негативные результаты ПЦР при скрининге и отсутствие повышенной температуры во время введения первой дозы вакцины, появление первых симптомов COVID-19 (4–5 дней после первой дозы, что сходно со средним инкубационным периодом COVID-19) свидетельствует о том, что участники были инфицированы SARS-CoV-2 до или около дня вакцинации, что было дополнительно подтверждено независимой наблюдательной комиссией на основе рекомендаций Всемирной организации здравоохранения (ВОЗ) и Центра по контролю и профилактике заболеваний США (CDC) и обзоре соответствующих клинических данных. Во-вторых, оба участника самостоятельно принимали нестероидные противовоспалительные средства, не поставив в известность врачей, что препятствовало постановке диагноза и получению медицинской помощи при госпитализации. В-третьих, в связи с ограничениями диагностики при скрининге (ограничения медицинского осмотра и тестирования, а также неосведомленность пациентов о наличии коморбидных патологий) у каждого пациента были коморбидные патологии, о которых стало известно только после госпитализации. У участников, которые не развили защитный иммунитет против SARS-CoV-2 (т.е. были инфицированы до вакцинации или в первое время после вакцинации), клиника течения COVID-19 была естественной. Мы провели дополнительный анализ серьёзности течения COVID-19 в двух группах, который показал, что в течение первых двух недель после введения первой дозы значимой разницы по тяжести течения COVID-19 между группами вакцины и плацебо не было. С 15-го по 21-й день после введения первой дозы эффективность составила 73,6% (p=0,048), затем, после 21-го дня, 100% (p<0,0001; приложение, с. 11). Поэтому в данном исследовании анализ эффективности проведён по данным через 21 день после введения первой дозы, поскольку к этому моменту формируется иммунный ответ.
В данный момент продолжается тщательный мониторинг, в особенности для учета случаев of COVID-19. Все данные по безопасности будут предоставлены регулирующим органам для анализа.
В этом промежуточном анализе у нас не было возможности оценить длительность защиты; медианное время наблюдения составило 48 дней после первой дозы. Хотя исследование включало участников с коморбидными патологиями, были представлены не все группы риска. Необходимы дальнейшие исследования вакцины у подростков и детей в соответствии с Планом педиатрических исследований, а также у беременных и кормящих женщин. Большинство участников данного клинического испытания принадлежали к белой расе, поэтому мы будем рады дальнейшим исследованиям в более разнообразных популяциях.
Промежуточные данные об эффективности были опубликованы для нескольких вакцин-кандидатов против SARS-CoV-2. Исследование безопасности и эффективности вакцины ChAdOx1 nCoV-19 (AZD1222) освещает анализ данных четырех рандомизированных контролируемых испытаний в Бразилии, Южной Африке и Великобритании. В анализ первичной эффективности были включены 11 636 участников. Среди участников, которые получили две стандартные дозы, эффективность вакцины составила 62,1%, а среди участников, получивших низкую дозу и затем стандартную дозу — 90,0%; общая эффективность составила 70,4% (2).
BNT162b2, мРНК-вакцина, разработанная Pfizer/BioNTech, показала 95% эффективность против COVID-19 в многонациональном плацебо-контролируемом базовом исследовании эффективности с ослеплением наблюдателей (4). В первичный анализ эффективности были включены 36 523 участника без инфекции в начале испытания. В экспериментальной группе наблюдали 8 случаев COVID-19, наступивших не менее чем через 7 дней после второй дозы; в группе плацебо наблюдали 162 таких случая. В экспериментальной группе был один случай тяжелого течения COVID-19, наступивший не ранее чем через 7 дней после введения второй дозы BNT162b2 (4). Рандомизированное стратифицированное плацебо-контролируемое исследование вакцины mRNA-1273 с ослеплением наблюдателей включало 30 000 участников, 25% которых составляли люди 65 лет или старше. Промежуточные результаты этого исследования позволяют заключить, что эффективность составляет 94,1%, на основе 95 случаев бессимптомного COVID-19 среди участников: 90 в группе плацебо и 5 в экспериментальной группе (3). Результаты данного клинического испытания Гам-КОВИД-Вас сходны с результатами, полученными для других вакцин.
Стратегии вакцинации должны учитывать ряд потенциальных проблем, связанных с приоритетным доступом к вакцинам против COVID-19 в разных группах, надёжной оценкой рисков нежелательных явлений вакцинации в группах с увеличенным риском тяжелого течения COVID-19 (люди старшего возраста и пациенты с коморбидными патологиями), логистикой распределения вакцины (холодильная цепь поставки), достаточным покрытием иммунизацией и длительностью защитного иммунного ответа. В соответствии с профилями целевых продуктов ВОЗ для вакцин против COVID-19 (16), характеристики, необходимые для использования в экстренных случаях во время вспышки, включают эффективность не менее 50%, пригодность для использования у людей старшего возраста, необходимость вводить не более двух доз и защиту на протяжении 6 месяцев. Для получения информации о длительности защитного иммунного ответа после введения вакцины необходимы дальнейшие исследования вакцин-кандидатов. Тем не менее, результаты по эффективности и безопасности вакцин-кандидатов против COVID-19 пока внушают оптимизм.
Наш промежуточный анализ этого клинического испытания III фазы Гам-КОВИД-Вак показал многообещающие результаты. Одновременно с применением в многочисленных клинических испытаниях (в России, Беларуси, Объединённых Арабских Эмиратах и Индии) эта вакцина уже выпущена в России для использования населением, в основном группами риска, медицинскими работниками и учителями, и по состоянию на 23 января 2021 г. более двух миллионов доз Гам-КОВИД-Вак уже были использованы для вакцинации населения (фармакологическая настороженность и наблюдение за редкими нежелательными явлениями находятся под контролем Федеральной службы по надзору в сфере здравоохранения).
Мы проводим исследование для изучение однодозового режима введения вакцины (клиническое испытание одобрено регулирующим этическим комитетом 8 января 2021 г., №1). Наш промежуточный анализ рандомизированного контролируемого испытания III фазы Гам-КОВИД-Вак в России показал высокую эффективность, иммуногенность и хорошую переносимость у участников от 18 лет.
Вклад соавторов
DYL — руководитель исследования, провёл исследование и координировал работу. IVD, DVS, AIT и AVK написали рукопись. IVD, NLL, YVS и EAT координировали исследование. IVD, OVZ, AIT, ASD, DMG, ASE, AVK, AGB, FMI, OP, TAO, IBE, IAF, DIZ, DVV, DNS и ASS собрали данные. IVD, OVZ, AIT, YVS, EAT, NLL, DAE, NAN, MMS и VAG внесли вклад в анализ и интерпретацию данных. DYL, IVD, DVS, SVB, BSN и ALG редактировали рукопись. IVD и DVS провели статистический анализ. EAS и SKZ: участие в организации, координации, проведении и технической поддержке исследования. ALG: участие в организации исследования и окончательном решении о публикации. Все авторы критически оценили рукопись и одобрили окончательную версию. Все авторы имеют полный доступ ко всем данным исследования и несут окончательную ответственность за решение о подаче статьи для публикации.
Заявление о наличии / отсутствии заинтересованности
OVZ, TAO, IVD, OP, DVS, DMG, ASD, AIT, DNS, IBE, EAT, AGB, ASE, ASS, SVB, DYL, BSN и ALG сообщают о наличии патентов на иммунобиологический экспрессионный вектор, фармацевтический агент и его использование для предотвращения COVID-19. Все остальные авторы заявляют об отсутствии конфликта интересов.
Ссылки
- WHO Draft landscape of COVID-19 candidate vaccines. (Проект ВОЗ вакцин-кандидатов COVID-19)
who.int - Voysey M, Clemens SAC, Madhi SA et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. (Безопасность и эффективность вакцины ChAdOx1 nCoV-19 (AZD1222) против SARS-CoV-2: промежуточный анализ четырех рандомизированных контролируемых исследований в Бразилии, Южной Африке и Великобритании)
Lancet. 2021; 397: 99-111. Full Text - Baden LR, El Sahly HM, Essink B et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. (Эффективность и безопасность вакцины mRNA-1273 SARS-CoV-2) N Engl J Med. 2020; (published online Dec 30.) doi.org
- Polack FP, Thomas SJ, Kitchin N et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. (Безопасность и эффективность mRNA вакцины BNT162b2 COVID-19) N Engl J Med. 2020; 383: 2603-2615. Google Scholar
- Zhang C, Zhou D. Adenoviral vector-based strategies against infectious disease and cancer. Hum Vaccin Immunother. 2016; 12: 2064-2074. Google Scholar. (Стратегии борьбы с инфекционными заболеваниями и раком, основанные на аденовирусных векторах. Hum Vaccin Immunother).
- Wold WS, Toth K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr Gene Ther. 2013; 13: 421-433. Google Scholar. (Аденовирусные векторы для генной терапии, вакцинации и генной терапии рака).
- Volpers C, Kochanek S. Adenoviral vectors for gene transfer and therapy. J Gene Med. 2004; 6: S164-S171. Google Scholar. (Аденовирусные векторы для переноса генов и терапии).
- Tatsis N, Ertl HC. Adenoviruses as vaccine vectors. Mol Ther. 2004; 10: 616-629. Full Text. (Аденовирусов в качестве векторов вакцины).
- Dolzhikova IV, Zubkova OV, Tukhvatulin AI, et al. Safety and immunogenicity of GamEvac-Combi, a heterologous VSV- and Ad5-vectored Ebola vaccine: an open phase I/II trial in healthy adults in Russia. Hum Vaccin Immunother. 2017; 13: 613-620. Google Scholar. (Безопасность и иммуногенность гетерологичной VSV-и Ad5 - векторизованной вакцины против Эболы GamEvac-Combi: открытое исследование I/II фазу здоровых взрослых в России).
- Lu S. Heterologous prime-boost vaccination. Curr Opin Immunol. 2009; 21: 346-351. Google Scholar. (Гетерологичная Прайм-буст вакцинация).
- Kovyrshina AV, Dolzhikova IV, Grousova DM et al. A heterologous virus-vectored vaccine for prevention of Middle East respiratory syndrome induces long protective immune response against MERS-CoV. Immunologiya. 2020; 41 (in Russian).: 135-143. Google Scholar. (Гетерологичная вирусовекторная вакцина для профилактики ближневосточного респираторного синдрома индуцирует длительный защитный иммунный ответ против MERS-CoV).
- Logunov DY, Dolzhikova IV, Zubkova OV et al. Safety and immunogenicity of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine in two formulations: two open, non-randomised phase 1/2 studies from Russia. Lancet. 2020; 396: 887-897. Full Text. (Безопасность и иммуногенность гетерологичной вакцины prime-boost COVID-19 на основе векторов rAd26 и rAd5 в двух препаратах: два открытых нерандомизированных исследования I/II фаз из России).
- Altman DG. Practical statistics for medical research. Chapman and Hall, London 1991. Google Scholar. (Практическая статистика медицинских исследований).
- WHO. Transmission of SARS-CoV-2: implications for infection prevention precautions.
who.int. (ВОЗ. Передача SARS-CoV-2: последствия для мер предосторожности по профилактике инфекции). - Centers for Disease Control and Prevention. Interim clinical guidance for management of patients with confirmed coronavirus disease (COVID-19). Updated Dec 8, 2020. (Центры по контролю и профилактике заболеваний. Промежуточное клиническое руководство по ведению пациентов с подтвержденным коронавирусным заболеванием (COVID-19).
cdc.gov. - WHO. Target product profiles for COVID-19 vaccines. who.int. (ВОЗ. Профили целевых продуктов для вакцин COVID-19).